Our Wholly Owned Clinical Product Pipeline
* Site specific protocol amendment with MD Anderson Cancer Center
** Bladder, Melanoma, Head & Neck, Ovarian, NSCLC, Esophageal, Gastric, Synovial sarcoma, MRCLS (myxoid/round cell liposarcoma)
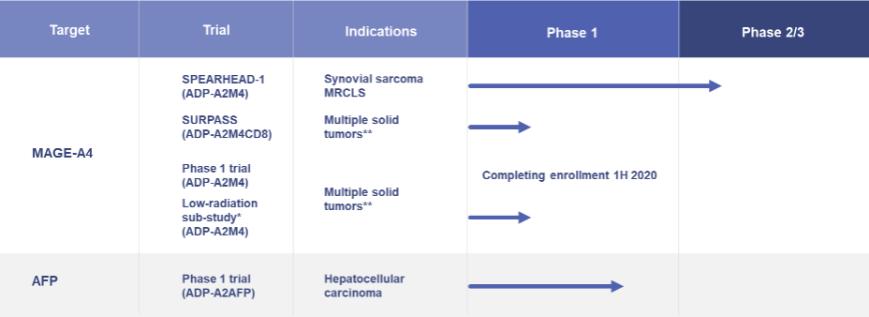
ADP-A2M4—Multiple Indications: Clinical trials are ongoing with our ADP-A2M4 SPEAR T-cell in multiple indications. In addition, planning is ongoing for initiation of a clinical trial combining ADP-A2M4 with a PD-1 / PD-L1 pathway inhibitor in 2020.
| ● | A Phase 1 clinical trial in multiple tumor indications, namely urothelial, melanoma, head and neck, ovarian, NSCLC, esophageal and gastric cancers, synovial sarcoma and MRCLS completed enrollment in early 2020. |
| o | As of October 23, 2019, data from 14 evaluable patients with synovial sarcoma treated in the expansion phase of this trial demonstrated an overall response rate of 50% (including both confirmed and unconfirmed partial responses (PRs)). 13 out of 14 evaluable patients had evidence of disease control (with best overall responses of partial response (7 patients) or stable disease (6 patients)). A clinical update was provided at the Connective Tissue Oncology Society in November 2019. |
| o | Beyond synovial sarcoma tumor shrinkage has been observed in patients with melanoma and ovarian cancers and a partial response was reported in a head and neck cancer patient. |
| ● | A Phase 2 clinical trial has been initiated in synovial sarcoma and MRCLS (“Spearhead -1”). The trial will take place at sites in the United States, Canada and Europe. The trial will include up to 60 patients at a selected dose of up to 10 billion transduced ADP-A2M4 SPEAR T-cells. Primary responses will be assessed by overall response rate by RECIST v1.1 (“Response Evaluation Criteria In Solid Tumors v1.1”). The lymphodepletion regimen will be fludarabine (30 mg/m2/day) for 4 days and cyclophosphamide (600 mg/m2/day) for 3 days. |
| ● | A radiation sub-study under the Phase 1 clinical trial is continuing at the MD Anderson Cancer Center. The sub-study will treat up to 10 patients and has a primary endpoint of safety, with RECIST v1.1 responses being a secondary endpoint. The radiation is a low dose radiation and is administered to lesions or isocenters prior to lymphodepletion. |
ADP-A2AFP - Hepatocellular Carcinoma: We continue dosing patients in our Phase 1, open-label, dose-escalation study designed to evaluate the safety and anti-tumor activity of our alpha fetoprotein (“AFP”) therapeutic candidate for the treatment of hepatocellular carcinoma, or HCC. The trial is open in the United States, United Kingdom
4