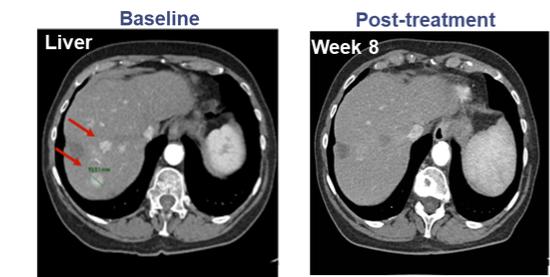
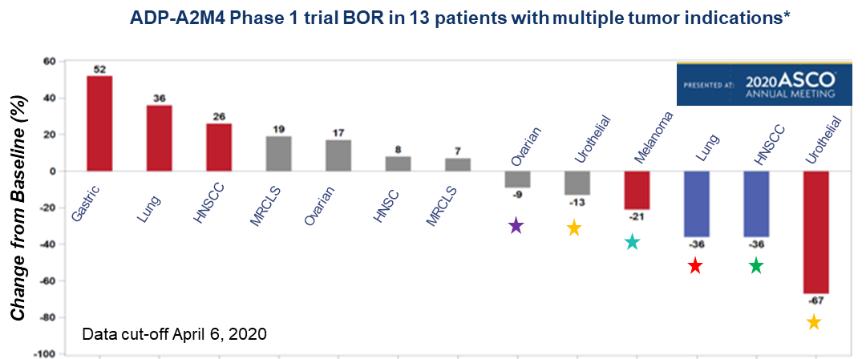
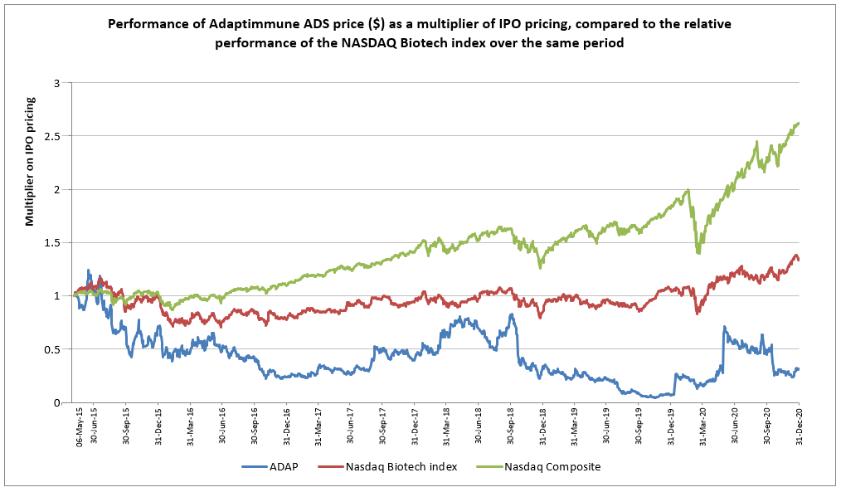
Government Regulation and Product Approvals
Government authorities in the United States, at the federal, state and local level, and in other countries and jurisdictions, including the EU and U.K, extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing, post-approval monitoring and reporting, and import and export of pharmaceutical products. The processes for obtaining regulatory approvals in the United States and in foreign countries and jurisdictions, along with subsequent compliance with applicable statutes and regulations and other regulatory authorities, require the expenditure of substantial time and financial resources. Failure to comply with the various federal, state and local level laws and requirements can also result in severe penalties and restrictions to the business.
FDA Approval Process
In the United States, therapeutic products, including drugs, biologics, and medical devices are subject to extensive regulation by the FDA. The Federal Food, Drug, and Cosmetic Act (the “FDC Act”), and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. Some biological products are subject to regulation under the FDC Act. Most biological products are approved for marketing under provisions of the Public Health Service Act (“PHSA”) via a BLA. The application process and requirements for approval of BLAs are generally similar to those for new drug applications (“NDAs”), and biologics are associated with generally similar, if not greater, approval risks and costs as drugs. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending NDAs or BLAs, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties, and criminal prosecution.
Biological product development for a new product or certain changes to an approved product in the United States typically involves preclinical laboratory and animal tests, the submission to the FDA of an Investigational New Drug application (“IND”), which must become effective before human clinical testing may commence, and adequate and well-controlled clinical trials to establish the safety and effectiveness of the drug for each indication for which FDA approval is sought. Satisfaction of FDA pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity, and novelty of the product or disease.
The FDA may order the temporary, or permanent, discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial either is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. An Investigational Review Board (“IRB”) may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions.
Clinical trials to support BLAs for marketing approval are typically conducted in three sequential phases, but the phases may overlap. In Phase 1, the initial introduction of the biologic into healthy human subjects or patients, the product is tested to assess metabolism, pharmacokinetics, pharmacological actions, side effects associated with increasing doses, and, if possible, early evidence on effectiveness. Phase 2 usually involves trials in a limited patient population to determine the effectiveness of the drug or biologic for a particular indication, dosage tolerance, and optimum dosage, and to identify common adverse effects and safety risks. If a compound demonstrates evidence of effectiveness and an acceptable safety profile in Phase 2 evaluations, Phase 3 trials are undertaken to obtain the additional information about clinical efficacy and safety in a larger number of patients, typically at geographically dispersed clinical trial sites, to permit the FDA to evaluate the overall benefit-risk relationship of the drug or biologic and to provide adequate information for the labeling of the product.
After completion of the required clinical testing, a BLA is prepared and submitted to the FDA. FDA approval of the BLA is required before marketing of the product may begin in the United States. The BLA must include the results of all preclinical, clinical, and other testing, compilation of data relating to the product’s pharmacology, chemistry, manufacture, and controls as well as proposed labeling for the product. The FDA has 60 days from its receipt